Dr. Tanja Domke, Dr. Mira Meyer-Ács, Yuk Ting Phung, Dr. Marc Esser (veröffentlicht in Market Access & Health Policy 2021; 11(3): 26–28)
Besondere Herausforderungen beim Market Access
Gentherapeutika in der frühen Nutzenbewertung
Mit der Entwicklung von Gentherapeutika ist die Zukunftsvision einer erfolgreichen Behandlung seltener, genetisch bedingter Erkrankungen Realität geworden. Die Verfügbarkeit neuartiger Therapieoptionen bietet zwar Heilungsaussichten für die betroffenen Patienten, stellt aber auch das Gesundheitssystem vor große Herausforderungen. Dies zeigt sich in verschiedenen Phasen des Markteintritts beginnend bei regulatorischen Besonderheiten bis hin zur Erstattung.
Was versteht man unter Gentherapie?
Erworbene oder angeborene Fehlfunktionen auf Ebene der Gene sind ursächlich für viele Krankheiten. Ist die genetische Ursache einer Erkrankung bekannt, so kann diese gezielt mittels Gentherapie behandelt werden. Im Gegensatz zu herkömmlichen Wirkstoffen, die Symptome einer Erkrankung pharmakologisch behandeln, wirken Gentherapeutika direkt auf die zelluläre Krankheitsursache ein. Ziel der Anwendung von Gentherapeutika ist allgemein die Heilung eines Gendefekts. Die aus einem solchen Defekt resultierenden Erkrankungen sind häufig selten, verlaufen allerdings für die Patienten meist chronisch, was die Bedeutung erfolgreicher Therapieoptionen hervorhebt.
Definition Gentherapeutikum
»Ein Gentherapeutikum ist ein biologisches Arzneimittel, dessen Wirkstoff eine Nukleinsäure (Träger der Erbinformationen) enthält oder daraus besteht. Es wird eingesetzt, um eine Nukleinsäuresequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu entfernen. Die therapeutische, prophylaktische oder diagnostische Wirkung steht in unmittelbarem Zusammenhang mit der rekombinanten Nukleinsäuresequenz, die es enthält oder mit dem Produkt, das auf Basis dieser genetischen Information gebildet wird.«
Quelle: Paul-Ehrlich-Institut1
Zulassung, Überwachung und Rückverfolgbarkeit
Gentherapeutika gehören neben den somatischen Zelltherapeutika und den biotechnologisch bearbeiteten Gewebeprodukten zu den Arzneimitteln für neuartige Therapien (Advanced therapy medicinal products, ATMPs). Zudem handelt es sich in den meisten Fällen um Arzneimittel für seltene Leiden (Orphan drugs), für die ebenfalls besondere Regelungen bei der Zulassung gelten. Alle modernen Arzneimittel der Biotechnologie müssen für die Zulassung in Europa das zentralisierte Zulassungsverfahren durchlaufen, welches von der EMA koordiniert wird. Dies gilt grundsätzlich auch für ATMPs und die dazu zählenden Gentherapeutika.
Die Regularien für ATMPs sind im Vergleich zu konventionellen Arzneimitteln umfangreich und stellen die Pharmaunternehmen vor besondere Herausforderungen. Beispielsweise hat der Antragsteller im Zulassungsantrag ausführlich die Maßnahmen zu erläutern, die er vorgesehen hat, um die Nachbeobachtung der Wirksamkeit und der Nebenwirkungen von ATMPs zu gewährleisten. Bei besonderem Anlass zur Besorgnis stellt die Kommission die Bedingung, dass ein Risikomanagementsystem eingerichtet wird oder dass der Zulassungsinhaber nach Markteinführung zusätzliche klinische Studien durchführt und der EMA zur Prüfung vorlegt. Ein weiterer Schwerpunkt der Regulierung liegt auf der Rückverfolgbarkeit. Diese muss für jedes einzelne Arzneimittel und seine Ausgangs- und Rohstoffe, einschließlich aller mit den möglicherweise darin enthaltenen Geweben oder Zellen in Berührung kommenden Stoffe gewährleistet sein. Die gesammelten Daten sind vom Zulassungsinhaber für mindestens 30 Jahre nach dem Verfallsdatum des Arzneimittels aufzubewahren. Auch das Krankenhaus, die Einrichtung oder die private Praxis, in dem/der das ATMP verwendet wird, hat ein System zur Rückverfolgbarkeit von Patienten und Arzneimitteln zu betreiben. Die EMA kann den Antragsteller oder Genehmigungsinhaber zur Pharmakovigilanz beraten.
Entwicklungsanreize
Die gute Nachricht: Die EMA schafft spezielle Anreize für die Entwicklung und die Marktzulassung von ATMPs. Die Gebühren für die wissenschaftliche Beratung in Bezug auf ATMPs sind um 90 % für kleine und mittlere Unternehmen sowie für andere Antragsteller um 65 % ermäßigt. Auch die Zulassungsgebühr wird um 50 % ermäßigt, wenn der Antragsteller ein Krankenhaus oder ein kleines oder mittleres Unternehmen ist und nachweisen kann, dass innerhalb der Gemeinschaft ein besonderes gesundheitsbezogenes Interesse an dem betroffenen ATMP besteht.2
Abseits des Zulassungsverfahrens durch die EMA ist es einzelnen Mitgliedstaaten der EU auch möglich, ATMPs auch auf nationaler Basis zu genehmigen. In Deutschland liegt die Zuständigkeit hierfür beim Paul-Ehrlich-Institut (PEI). Bislang wurden allerdings alle auf dem deutschen Markt verfügbaren Gentherapeutika im zentralisierten Verfahren zugelassen.3
Frühe Nutzenbewertung und anwendungsbegleitende Datenerhebung
Gentherapeutika können durch eine einmalige Anwendung zur Heilung einer zumeist seltenen, oftmals lebensbedrohlichen Erkrankung führen. Dieser revolutionäre Ansatz stellt besondere methodische Herausforderungen an die frühe Nutzenbewertung: ein Zusatznutzen muss an wenigen Patienten, bei wenigen Therapiealternativen über einen langen Zeithorizont beurteilt werden. Klinische Studien als Bewertungsgrundlage für die Zulassung sind häufig aufgrund der geringen Patientenzahlen einarmig. Randomisierte, aktiv kontrollierte Studiendesigns, wie sie für die Bewertung des Zusatznutzens gefordert werden, sind schwer darstellbar, denn aufgrund der zumeist wenigen Therapiealternativen ist die Festsetzung einer aktiven Vergleichstherapie schwierig. Hier greift zunächst die Orphan Drug-Regelung der frühen Nutzenbewertung: Es wird mit der Zulassung ein nicht quantifizierbarer Zusatznutzen zuerkannt, und die Festlegung einer zweckmäßigen Vergleichstherapie entfällt. Wird allerdings eine Umsatzgrenze von 50 Mio. € pro Jahr überschritten, wird auch für ein Orphan Drug eine zweckmäßige Vergleichstherapie bestimmt. Nun entsteht die Schwierigkeit, dass für einen Vergleich der Jahrestherapiekosten die einmalige Gabe der Gentherapie gegen eine kontinuierliche Therapie aufgerechnet werden muss. Ein weiteres Problem ist der Zeithorizont: Gentherapien sind durch die Integration der Genkorrektur in das Patientengenom zumindest theoretisch lebenslang wirksam. Die Langzeiteffekte der Gentherapie sind innerhalb des in der Nutzenbewertung dargestellten Zeithorizonts von mehreren Monaten bis hin zu wenigen Jahren nicht zu erfassen und können deswegen nicht abschließend bewertet werden.
Als Reaktion auf die häufig magere Datenlage bei der Zulassung und in der frühen Nutzenbewertung von Orphan Drugs wie auch von Arzneimitteln mit bedingter Zulassung und Arzneimitteln mit Zulassung unter außergewöhnlichen Umständen („exeptional circumstances“) wurde vom Gesetzgeber die anwendungsbegleitende Datenerhebung (abD) eingeführt. Diese verpflichtet den pharmazeutischen Unternehmer dazu, Daten aus der klinischen Routine zu erheben und diese für eine Zusatznutzenbewertung auszuwerten. Zum ersten Mal hat der G-BA im Februar 2021 eine abD für das Arzneimittel Zolgensma® angefordert und damit einen Präzedenzfall geschaffen. Zolgensma®, eine Gentherapie zur Behandlung der seltenen Erkrankung Spinale Muskelatrophie, war von der EMA bedingt zugelassen worden. Nach Überschreiten der 50 Mio. €-Umsatzgrenze wurde im Rahmen der abD mit Spinraza® eine zweckmäßige Vergleichstherapie bestimmt, und es wurde eine Berichtspflicht in Form eines Dossiers nach einem definierten Zeitraum der Datenerhebung festgelegt. Der Einsatz der Gentherapie wurde außerdem auf an der Datenerhebung mitwirkende medizinische Zentren begrenzt, so wie es im Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) vorgesehen ist.
Es ist absehbar, dass die abD bei Gentherapeutika der Standard werden wird. In einem Beschluss vom 01. April 2021 beauftragte der G-BA das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) mit der Ausarbeitung eines Konzepts zur abD für eine Wirkstoffklasse, und zwar am Beispiel der CAR-T-Zelltherapien, einer besonderen Form der Gentherapie. Insbesondere sollen die Art, Dauer und der Umfang der Datenerhebung konkretisiert werden. Daneben sollen eine Fragestellung einschließlich der zu erfassenden patientenrelevaten Endpunkte, die Methodik der Datenerhebung und die Datenauswertung erarbeitet werden. Auch die bestehenden Therapiealternativen sollen betrachtet und relevante Wirkstoffklassen bzw. Therapieansätze identifiziert werden. Dieses Konzept soll dem G-BA zum 30. September 2021 vorliegen.4
Für die Hersteller von Gentherapien bedeutet die abD, dass neue strategische Erwägungen notwendig werden. Kann ein Marktzugang mittels einer bedingten Zulassung zukünftig leichter erreicht werden, wenn im Nachgang mit einer abD zu rechnen ist? Wie hoch werden Aufwand und Kosten der abD für das pharmazeutische Unternehmen? Können die Kosten der abD durch einen frühen Marktzugang ausgeglichen werden? Es bleibt weiterhin zu beobachten, wie häufig der G-BA von der Möglichkeit der Forderung einer abD Gebrauch machen wird und wie umfangreich diese ausgestaltet werden wird.
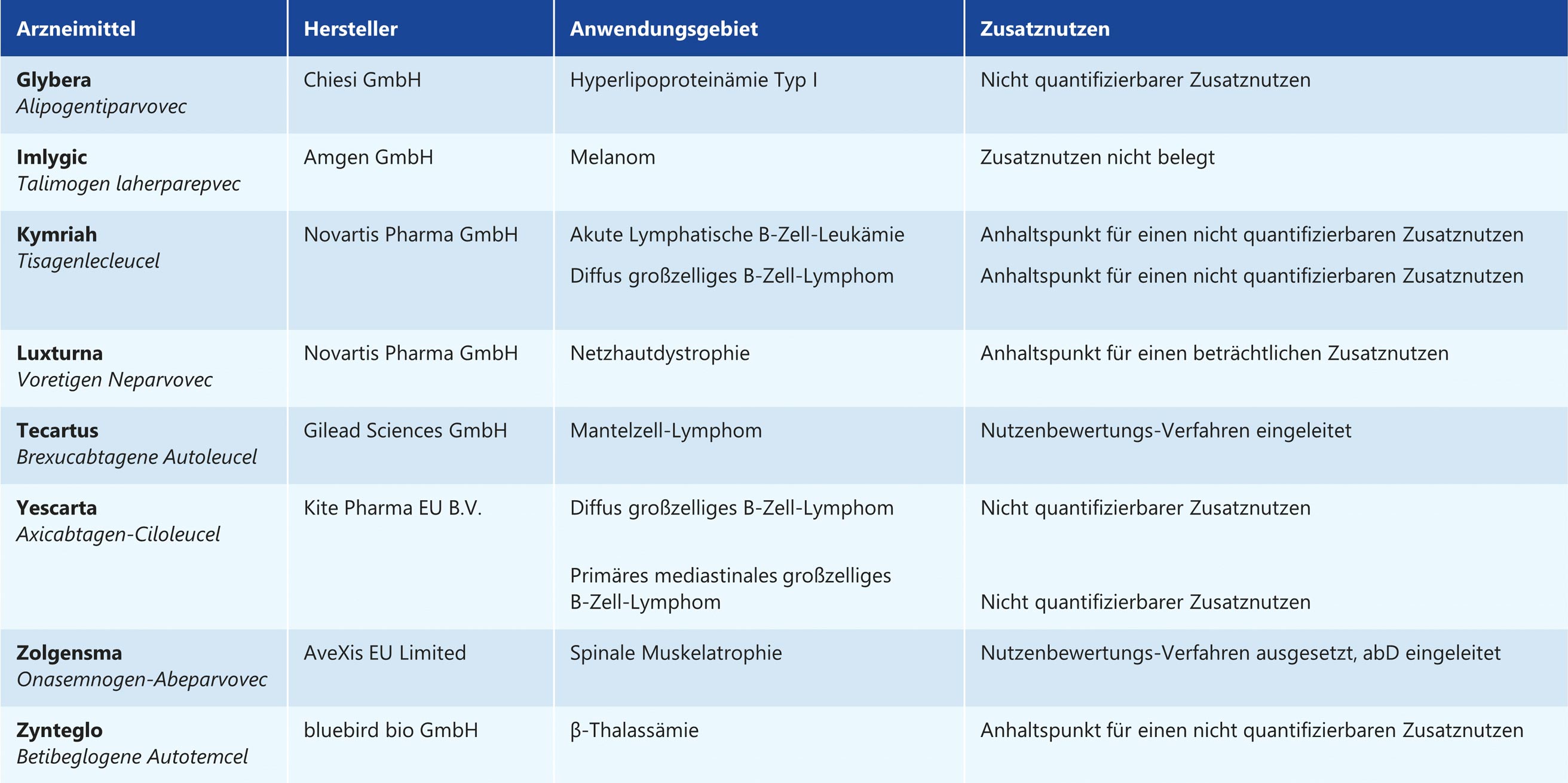
Tabelle 1: Gentherapeutika in der frühen Nutzenbewertung, Stand 12.04.2021
Erstattung von Gentherapeutika
Eine weitere Besonderheit der Gentherapeutika liegt in deren Erstattung. Da diese Medikamente in der Regel auf eine einmalige Gabe abzielen, die dann im Idealfall zur Heilung der jeweiligen Erkrankung führt, entstehen hohe einmalige Erstattungskosten. Die bisherige deutsche Erstattungslandschaft kann diese Anforderungen nur sehr bedingt abbilden. Haupteinsatzgebiet der meisten Gentherapeutika ist der stationäre Bereich, in dem die Erstattung innerhalb des DRG-Systems (Diagnosis Related Groups) erfolgt. Bislang besteht die einzige Möglichkeit für eine extrabudgetäre Vergütung eines höheren Arzneimittelpreises in der Anwendung des NUB-Verfahrens, welches die Kosten für neue Untersuchungs- und Behandlungsmethoden (NUB) abbilden soll.
Sowohl für die pharmazeutischen Unternehmen als auch für die Kostenträger gestaltet sich die faire Erstattung von Gentherapeutika in Deutschland als Gratwanderung. Der pharmazeutische Unternehmer muss seine hohen Kosten für Forschung und Entwicklung durch eine einmalige Erstattung decken. Zudem ist im Bereich der Orphan Drugs nur mit kleinen Patientenzahlen und daher wenigen Anwendungsfällen zu rechnen. Auf der anderen Seite sieht sich der Kostenträger mit sehr hohen Behandlungskosten zum Zeitpunkt der Verabreichung konfrontiert. Zwar existieren kaum Compliance-Probleme, die einem Therapieerfolg entgegenstehen könnten, jedoch kann nicht davon ausgegangen werden, dass der Behandlungserfolg nach mehreren Jahren oder Jahrzehnten anhält. Auch Folgekosten durch späte Nebenwirkungen stellen für den Kostenträger ein potentielles Risiko dar. So besteht ein dringender Bedarf an der Etablierung innovativer Erstattungsmodelle, die beiden Seiten gerecht werden.
Eine Möglichkeit sind hier erfolgsabhängige Verträge bzw. Zahlungsmodelle (Pay for Performance/Risk Sharing). Bis zu einem bestimmten Zeitpunkt müssen bei diesem Modell vordefinierte Behandlungserfolge nachweisbar sein. Ist dies nicht der Fall, so verpflichtet sich der pharmazeutische Unternehmer, den Erstattungsbetrag ganz oder teilweise an den Kostenträger zurück zu zahlen. Ein anderes Vertragsmodell sieht eine Art Ratenzahlung vor: Der Preis der Therapie wird über mehrere Jahre verteilt gezahlt. Auch diese Variante lässt sich direkt an den Behandlungserfolg koppeln, indem Zahlungen bei einem Rückfall oder Progress der Erkrankung eingestellt werden. Sollen Zahlungen an patientenrelevante Outcomes gebunden sein, so muss jedoch Einigkeit darüber erlangt werden, welche Parameter für die Evaluierung geeignet sind. Je nach Indikation kann dies eine weitere Hürde darstellen. Unklar bleibt dabei auch, wem die Bewertung und Überwachung der jeweiligen Outcome-Parameter obliegt. Gerade kleinere Krankenkassen werden solche Verträge kaum eigenständig monitoren können.
Ein viel diskutiertes Beispiel für die Etablierung eines alternativen Erstattungsmodells im deutschen Versorgungskontext bietet die CAR-T-Zelltherapie Kymriah® von Novartis. Hier wurde mit einem Teil der Krankenkassen vereinbart, dass ihnen die Therapiekosten teilweise zurückerstattet werden müssen, wenn der Patient oder die Patientin trotz Behandlung mit Kymriah® verstirbt. Für Yescarta® von Kite/Gilead wurde ein ähnlicher Outcome-orientierter Vertrag geschlossen. Auch hier richtet sich die Rabattierung nach dem Überleben der behandelten Patienten. Neustes Beispiel für ein innovatives Pay-for-Performance-Konzept ist die Gentherapie Zynteglo® von bluebird bio: Vorgesehen ist hier eine gestaffelte Zahlung über vier Jahre hinweg. Dabei werden 20 % der Gesamtsumme bei Verabreichung fällig, weitere 20 % ein Jahr später sofern die Behandlung angeschlagen hat. Nachfolgend wird der Therapieerfolg alle 12 Monate anhand eines vordefinierten Endpunkts erneut geprüft. Hält der Behandlungserfolg an, so sind dann die weiteren Raten zu zahlen.
Ausblick und Fazit
Die Heilung schwerwiegender Erkrankungen durch Gentherapeutika birgt große Chancen für die betroffenen Patienten, aber ebenso große Herausforderungen für das Gesundheitssystem und insbesondere das AMNOG-Verfahren. Die Zukunft der Gentherapeutika in Deutschland wird mit neuen Anforderungen zur Datenerhebung verbunden sein, wobei von einer zunehmenden Bedeutung der abD und einer darauf basierenden nachgelagerten Nutzenbewertung ausgegangen werden kann. Auch die Etablierung ausgereifter Erstattungskonzepte wird weiterhin ein Problem bleiben, an deren Lösung pharmazeutische Unternehmer und Kostenträger gemeinsam arbeiten müssen. Mit zunehmender Anzahl von Gentherapeutika stellt sich besonders bei Pay-for-Performance-Erstattungsmodellen auch die Frage nach der Notwendigkeit eines zentralen Monitorings, um den Therapieerfolg im Hinblick auf Zahlungsmeilensteine unabhängig zu bewerten. Neue Entwicklungen im Bereich des AMNOG sind in den kommenden Jahren im Bereich der Orphan Drugs zu erwarten. Die Privilegien der Orphan Drugs werden von G-BA und Payern zunehmend kontrovers diskutiert. Zentral ist hierbei die Kostenfrage. Künftige Anpassungen des AMNOG-Verfahrens werden wahrscheinlich darauf abzielen, die Orphan Drugs ihres Sonderstatus zumindest teilweise zu entheben, um so die Kosten für diese Präparate zu reduzieren. Da die meisten Gentherapien gegenwärtig zu den Orphan Drugs zählen, wird sich das unmittelbar auch auf die Gentherapien auswirken.
Literatur
1 Paul-Ehrlich-Institut,. Gentherapeutika. [Online] [Zitat vom: 11. 03 2021.] https://www.pei.de/DE/arzneimittel/atmp/gentherapeutika/gentherapeutika-node.html;jsessionid=A05611A616EE879E0E282FC2847C374A.intranet212.
2 Amtsblatt der Europäischen Union. VERORDNUNG (EG) Nr. 1394/2007 . [Online] [Zitat vom: 06. 04 2021.] https://eur-lex.europa.eu/legal-content/DE/TXT/PDF/?uri=CELEX:32007R1394&from=de.
3 Paul-Ehrlich-Institut. Arzneimittel für neuartige Therapien. [Online] [Zitat vom: 06. 04 2021.] https://www.pei.de/SharedDocs/Downloads/DE/regulation/beratung/innovationsbuero/broschuere-atmp.pdf?__blob=publicationFile&v=4.
4 Gemeinsamer Bundesausschuss. Gemeinsamer Bundesausschuss: Pressemitteilungen und Meldungen. AMNOG: G-BA vergibt Aufträge ans IQWIG – Konzeptentwicklung und Nutzenbewertung. [Online] 01. April 2021. [Zitat vom: 07. April 2021.] https://www.g-ba.de/presse/pressemitteilungen-meldungen/948/.