Dr. Mira Meyer-Ács, Laura Loke, Dr. Marc Esser (veröffentlicht in Market Access & Health Policy 2022; 12(4): 22–24)
Die europäische Nutzenbewertung für Arzneimittel kommt – was bisher bekannt ist
Mit der Verabschiedung der EU-HTA-Verordnung ist der Europäischen Kommission ein großer Wurf gelungen: Sie bezieht sich auf alle Gesundheitstechnologien auf dem europäischen Markt und definiert, welche Arzneimittel, Medizinprodukte und In-vitro-Diagnostika zukünftig gemeinsam auf europäischer Ebene klinisch bewertet werden müssen. Wir blicken auf die möglichen Auswirkungen der Verordnung und ihrer Konkretisierung durch EUnetHTA 21 auf die Nutzenbewertung von innovativen Arzneimitteln in Deutschland.
Hintergrund
Seit Jahrzehnten wird die Idee einer europäischen Nutzenbewertung, also einer gemeinsamen Bewertung von Gesundheitstechnologien in der Europäischen Union (EU-HTA), diskutiert, um die Bewertung von Arzneimitteln und Medizinprodukten innerhalb der EU zu vereinheitlichen und die Bewertungsprozesse zu vereinfachen. Im Jahr 2006 organisierten sich 83 nationale Organisationen zum European Network for Health Technology Assessment (EUnetHTA), um die europäische Nutzenbewertung voranzutreiben. Mit der Richtlinie 2011/24/EU vom 09.03.2011 zur Ausübung der Patientenrechte in der grenzüberschreitenden Gesundheitsversorgung wurde der rechtliche Rahmen für die Unterstützung eines HTA-Netzwerks durch die EU geschaffen.
Der Gesetzgebungsprozess zur nun vorliegenden Verordnung (EU) 2021/2282 über die Bewertung von Gesundheitstechnologien und zur Änderung der Richtlinie 2011/24/EU war langwierig und von einem intensiven Ringen um Kompromisse gezeichnet: Bereits 2018 wurde ein erster Entwurf vorgestellt, die finale Fassung wurde jedoch erst am 22.12.2021 im Amtsblatt der EU-Kommission veröffentlicht.1
Die Ziele der EU-HTA-Verordnung sind klar gesteckt. Im Zentrum steht die Förderung von Innovationen: Durch die gemeinsame Bewertung sollen bestmögliche Ergebnisse für Patienten und die Gesellschaft erzielt werden. Gleichzeitig sollen Hersteller und Unternehmen entlastet werden. Insbesondere wird ein geringerer bürokratischer Aufwand angestrebt, indem alle für die gemeinsame klinische Bewertung (Joint Clinical Assessment, JCA) relevanten Informationen und Daten nur einmal auf europäischer Ebene eingereicht werden müssen. Dies soll die Vielzahl von Bewertungsverfahren in den verschiedenen Ländern vereinfachen.
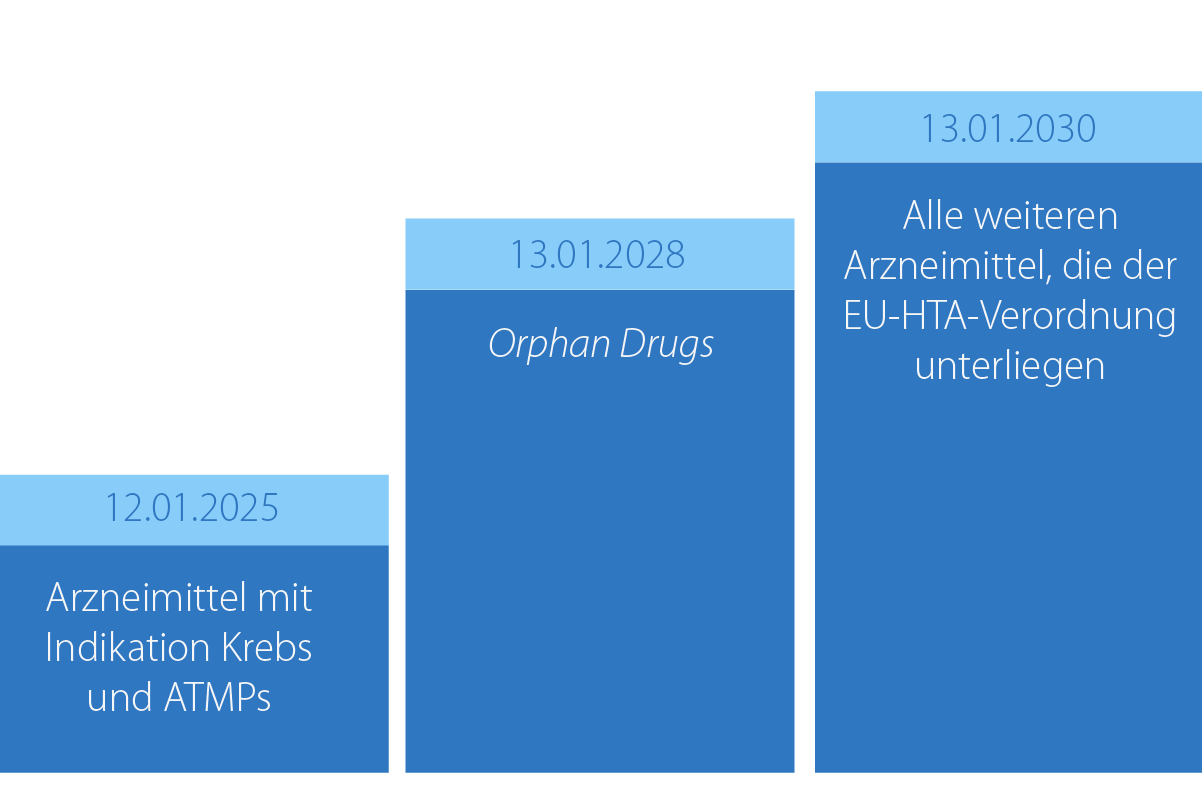
Abbildung 1: Die Einführung des JCA erfolgt stufenweise für verschiedene Arzneimittelgruppen. Dabei ist das Datum des Antrags auf Inverkehrbringen maßgeblich.
EUnetHTA 21
In den Jahren 2010 bis 2021 wurden im Rahmen der EUnetHTA Joint Actions 1–3 grundlegende Vorarbeiten geleistet, indem Konzepte zur Definition und Umsetzung eines nachhaltigen Modells für die wissenschaftliche und technische Zusammenarbeit bei der Bewertung von Gesundheitstechnologien in Europa entwickelt wurden.
Darauf aufbauend ist nun das Konsortium EUnetHTA 21 mit einem Dienstleistungsvertrag über die Durchführung gemeinsamer Arbeiten zur Bewertung von Gesundheitstechnologien beauftragt, um die Fortsetzung der EU-Zusammenarbeit im Bereich des Health Technology Assessment (HTA) zu unterstützen. Das Konsortium besteht aus 13 nationalen HTA-Organisationen, dazu gehören der Gemeinsame Bundesausschuss (G-BA) und das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG). EUnetHTA 21 ist für die Entwicklung der gemeinsamen wissenschaftlichen Beratungen (Joint Scientific Consultations, JSC) mit der European Medicines Agency (EMA) sowie für die Ausgestaltung der Prozesse und Methoden zur JCA im Einklang mit der EU-HTA-Verordnung zuständig. Für die methodischen Arbeitsergebnisse von EUnetHTA 21 werden öffentliche Konsultationen durchgeführt.2
Gegenstand der EU-HTA-Verordnung
Die EU-HTA-Verordnung bietet einen Unterstützungsrahmen für die Zusammenarbeit von Mitgliedsstaaten im Bereich der HTA auf Ebene der Europäischen Union. HTA ist als multidisziplinärer Prozess definiert, in den Informationen über medizinische, patientenbezogene und soziale Aspekte sowie wirtschaftliche und ethische Fragen einbezogen werden.
Ein besonderes Augenmerk liegt dabei auf der JCA: Durch das Verfahren soll sichergestellt werden, dass Informationen, Daten, Analysen und sonstige Nachweise für die klinische Bewertung nur einmal auf Unionsebene vorgelegt werden müssen, und dass die klinische Bewertung nach gemeinsamen Vorschriften und Methoden durchgeführt wird.
Explizit von der Verordnung nicht berührt ist die Zuständigkeit der Mitgliedsstaaten, Schlussfolgerungen über die relative Wirksamkeit von Gesundheitstechnologien zu ziehen oder Entscheidungen über den Einsatz einer Gesundheitstechnologie im spezifischen nationalen Gesundheitskontext zu treffen. Konkret zählen zu den Aufgaben der einzelnen Mitgliedsstaaten die nicht-klinische Bewertung, welche ökonomische, organisatorische und ethische Aspekte umfasst. Außerdem können bei Bedarf ergänzende klinische Analysen (z.B. aus nationalen Patientenregistern) bewertet werden. Schließlich ist es nationale Aufgabe, Schlussfolgerungen aus den Daten zu ziehen, um einen Beschluss über den Zusatznutzen im Kontext des nationalen Gesundheitswesens zu treffen.
Die EU-HTA-Verordnung wird für obligatorisch und optional zentral (über die EMA) zuzulassende Arzneimittel sowie auch für bereits gemeinsam klinisch bewertete Arzneimittel, die eine neue Indikation erhalten, gelten. Die Einführung des JCA ist stufenweise geplant (siehe Abbildung 1), zunächst für Arzneimitteln mit der Indikation Krebs und den Advanced Therapy Medicinal Products (ATMPs) im Jahr 2025. Ab 2030 wird sie für alle Arzneimittel, die in den Geltungsbereich der EU-HTA-Verordnung fallen, bindend.
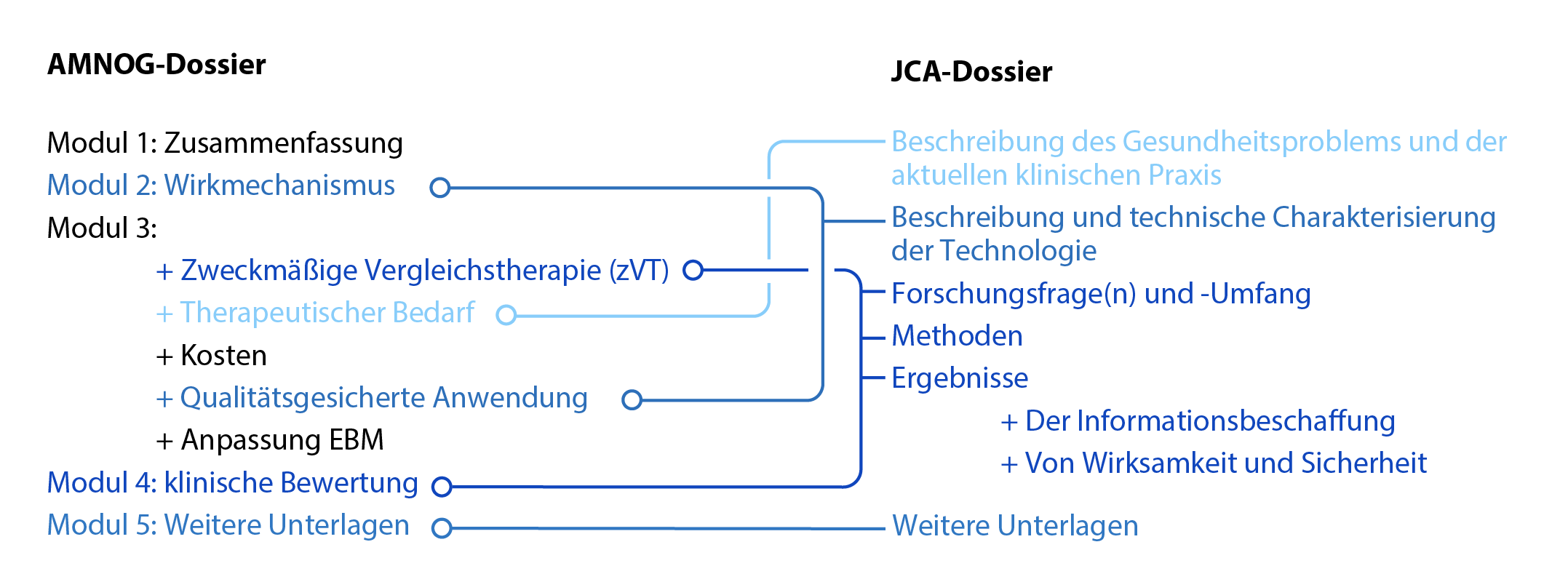
Abbildung 2: Inhaltliche Berührungspunkte zwischen AMNOG- und JCA-Dossier.
AMNOG- und JCA-Dossier – Berührungspunkte
Der Bewertungsumfang im Rahmen der JCA wird von den Mitgliedsstaaten gemeinsam auf Basis des Population Intervention Comparator Outcome (PICO)-Schema festgelegt. Neben der reinen Ergebnisdarstellung wird eine wissenschaftliche Analyse der klinischen Effekte (einschließlich Mortalität, Morbidität, Nebenwirkungen, gesundheitsbezogene Lebensqualität) und die Diskussion wissenschaftlicher Unsicherheiten gefordert. Während die erste Veröffentlichung der JCA-Dossiervorlage für Ende Juli 2022 erwartet wird, sind die übergeordneten Dossierinhalte in der EU-HTA-Verordnung definiert. Ein Vergleich zwischen den Anforderungen der EU-HTA-Verordnung und dem AMNOG-Nutzendossier zeigt, dass einige Bestandteile des JCA-Dossiers bestimmten Abschnitten des AMNOG-Dossiers entsprechen und diese ersetzen könnten (s. Abbildung 2). Beispielsweise weist die Beschreibung und technische Charakterisierung der Technologie im Falle der innovativen Arzneimittel einen Bezug zu Modul 2 des AMNOG-Dossiers auf. Es ist davon auszugehen, dass einige Teile des AMNOG-Dossiers künftig weniger umfangreich ausfallen, wenn ein Großteil der Unterlagen bereits auf europäischer Ebene im JCA-Dossier vorgelegt wird.
Erste Schwierigkeiten werden erkennbar, wenn die Darstellung des Gesundheitsproblems und der aktuellen klinischen Praxis im JCA-Dossier betrachtet wird, welches der Beschreibung des therapeutischen Bedarfs in Modul 3 des AMNOG-Dossiers nahekommt. Insbesondere die klinische Praxis kann sich in den einzelnen Mitgliedstaaten stark voneinander unterscheiden. Hier sind medizinische Leitlinien maßgeblich, deren europaweite Vereinheitlichung wünschenswert ist und zum Teil angestrebt wird. Bislang jedoch obliegt die Leitlinienerstellung in vielen Bereichen nationalen Institutionen.
Offen ist, wie umfangreich sich der Abschnitt zu Forschungsfrage(n) und -Umfang im JCA-Dossier gestalten wird. Hier bestehen Berührungspunkte mit mehreren Abschnitten des AMNOG-Dossiers, insbesondere Modul 4. Geplant ist, dass der Bewertungsumfang gemäß PICO-Schema in den einzelnen Mitgliedstaaten abgefragt und durch eine nachfolgende Konsolidierung auf die kleinstmögliche Anzahl unterschiedlicher Fragestellungen festgelegt wird. Es ist wahrscheinlich, dass der pharmazeutische Unternehmer in vielen Fällen die parallele Analyse mehrerer Forschungsfragen in einem JCA-Dossier liefern muss.
Zeitlicher Ablauf der gemeinsamen klinischen Bewertung
Problematisch für die pharmazeutischen Unternehmen erscheint dabei der geplante zeitliche Verfahrensablauf. Während das AMNOG-Verfahren nach der Zulassung und mit der Markteinführung des neuen Arzneimittels startet, soll die JCA zu einem sehr frühen Zeitpunkt parallel zum Zulassungsverfahren beginnen, wenn noch nicht eindeutig feststeht, ob es überhaupt eine Zulassung geben wird und wofür genau diese gilt. Konkret soll die Absichtserklärung (Letter of Intent) des pharmazeutischen Unternehmers bereits einige Tage bzw. Wochen nach dem Start des Zulassungsverfahrens eingereicht werden. Das Beratungstreffen (Scoping Meeting), in welchem der Bewertungsumfang festgelegt wird, ist ca. 100 Tage vor der Stellungnahme des Committee for Medicinal Products for Human Use (CHMP opinion) angesetzt (siehe Abbildung 3). Die Einreichung des JCA-Dossiers ist 45 Tage vor der CHMP opinion geplant, womit sich der Zeitraum für die Erstellung des Dossiers nach dem Scoping Meeting auf gerade einmal 55 Tage beläuft. Die finale Version des JCA soll ca. 30 Tage nach der Zulassung veröffentlicht werden, um die weitere Bewertung im nationalen Rahmen nicht zu verlangsamen. Nicht abschließend geklärt ist, wie im Rahmen des JCA damit umgegangen werden soll, wenn sich im Laufe des Zulassungsprozess das Anwendungsgebiet (Label) ändert. Wahrscheinlich ist, dass dies eine deutliche Verzögerung des JCA-Prozesses auslösen wird.
Zunächst wird ein Testlauf mit ein bis zwei JCA im Rahmen von EUnetHTA 21 durchgeführt. Die Ergebnisse sollen bis Sommer 2023 vorliegen.
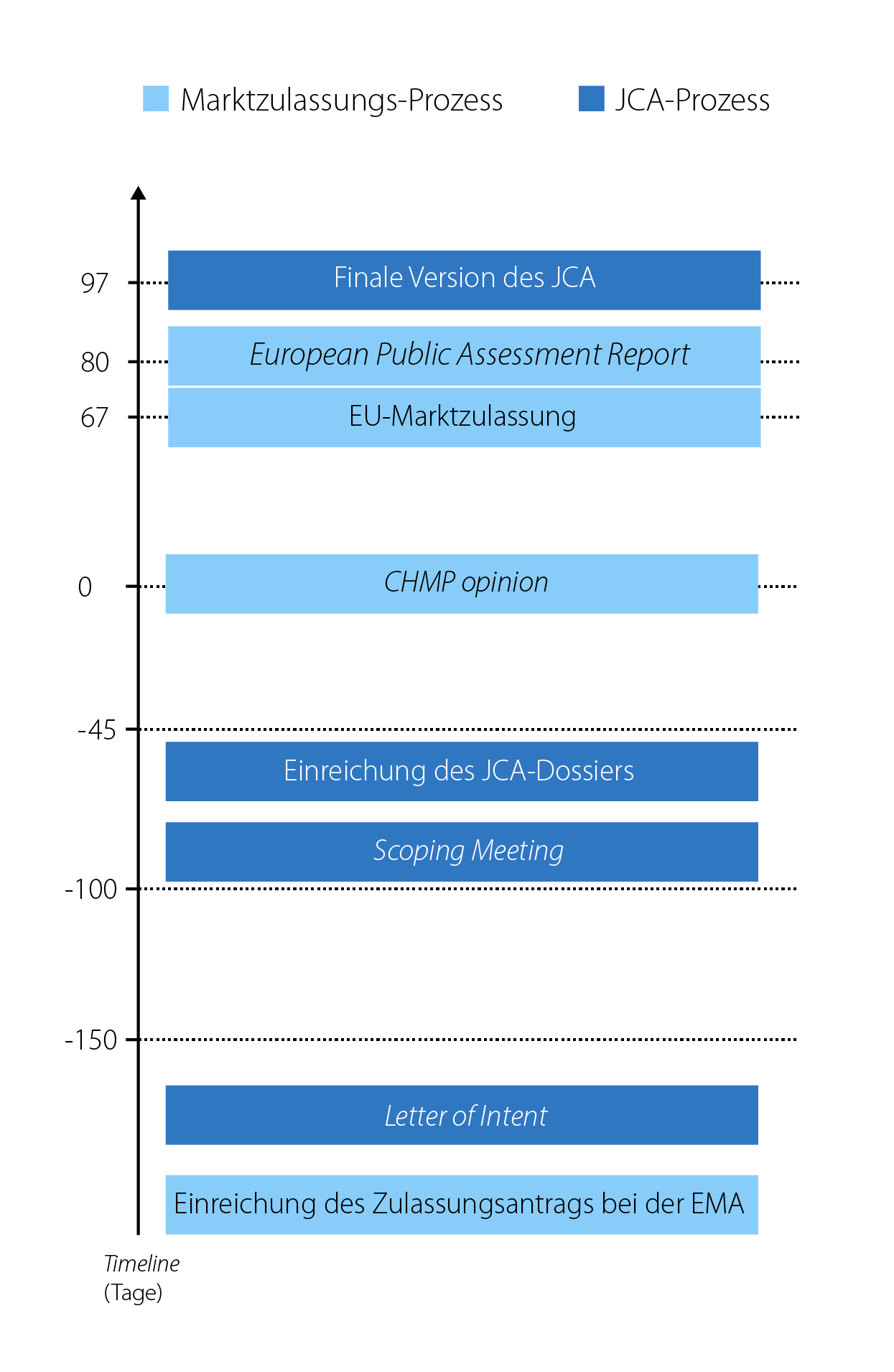
Abbildung 3: Zeitlicher Ablauf von EU-Marktzulassung und geplanter JCA (vereinfacht dargestellt).
Fazit
Die EU-HTA-Verordnung schafft neue Chancen, aber auch neue Herausforderungen für pharmazeutische Unternehmen. Während die gemeinsame klinische Bewertung zukünftig auf europäischer Ebene zentralisiert wird, bleibt die nationale Zuständigkeit für die Bewertung im landesspezifischen Versorgungskontext und die darauf basierende Ableitung eine Zusatznutzens bestehen. Das JCA-Dossier wird in vielen Punkten dem AMNOG-Dossier ähneln, sodass die tiefe Kenntnis des deutschen AMNOG-Verfahrens im Hinblick auf die europäische Nutzenbewertung von Vorteil sein dürfte. Insofern sollten auch deutsche Verantwortliche im Market Access und deutsche Dienstleister im Kontext der europäischen Nutzenbewertung weiterhin gefragt sein.
Quellen
1. EUR-Lex. Verordnung (EU) 2021/2282 des Europäischen Parlaments und des Rates vom 15. Dezember 2021 über die Bewertung von Gesundheitstechnologien und zur Änderung der Richtlinie 2011/24/EU (2021). Verfügbar unter: https://eur-lex.europa.eu/legal-content/DE/TXT/?uri=CELEX:32021R2282. Zugriffsdatum: 07.06.2022.
2. EUnetHTA. Public Consultation (2022). Verfügbar unter: https://www.eunethta.eu/public-consultation/. Zugriffsdatum: 07.06.2022.